|
Молекулярная динамика пептидов. |
К.В. Шайтан
|
В статье рассмотрены основы метода молекулярной динамики и его приложения к изучению динамики белков и пептидов. Обсуждаются структура энергетической поверхности и эргодичность траекторий. Излагаются представления о динамических корреляционных функциях, картах уровней свободной энергии, электронно-конформиационных взаимодействиях.
|
Введение.
Молекулярная
динамика биополимеров находится на стыке двух наиболее быстро развивающихся в
настоящее время областей науки – физико-химической биологии и информатики
(более точным является термин “computer
science”). Прогресс в компьютерных
технологиях за последние 20 лет оказался столь стремительным, что породил определенные
надежды при решении задач, которые казались ранее неразрешимыми. Так вполне
реальным становится создание систем по типу САПР (напомним, что в технике это
означает компьютерные «системы автоматического проектирования») для
проектирования и конструирования молекулярных устройств с заданными функциями.
Такие устройства могут быть биологической природы, как, например, молекулярные
моторы, обеспечивающие важнейшие клеточные функции [1], так и молекулярные
устройства не биологической, синтетической, природы. Эти устройства привлекают
в последнее время внимание не только писателей-фантастов, но и весьма солидные
фирмы, занимающиеся разработкой нанотехнологий [2].
Возможно, в ближайшем будущем возникнет и новая специальность, например,
«молекулярный инженер-конструктор». Было бы неправильно думать, что проблемы
при молекулярном моделировании достаточно сложных систем сводятся к
использованию как можно более мощной
компьютерной техники. Здесь ситуация может быть и обратной – чем мощнее
компьютер, тем больше артефактов возникает при малейшем пренебрежении
фундаментальными физическими свойствами молекул, которые, к сожалению, нам
известны далеко не полностью. Ниже, мы лишь очень кратко продемонстрируем суть
метода молекулярной динамики и возникающие здесь проблемы.
Уравнения
движения и атом-атомные
потенциалы.
Рассмотрим, например, фрагмент пептидной цепи
(рис.1).
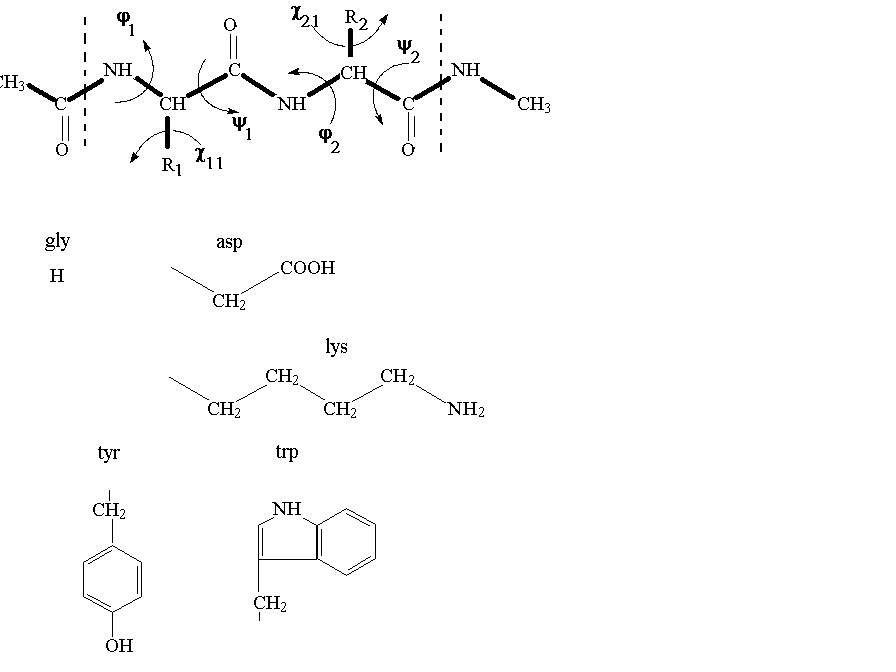
Рис.1. 1- структура фрагмента пептидной цепи и обозначения двугранных (торсионных) углов. При моделировании динамики пептидов для устранения концевых эффектов обычно с N – конца присоединяют ацетил, с С- конца – метиламин (отделены от основной цепи пунктирами). 2 – примеры боковых радикалов в природных аминокислотах: gly – глицин, asp – аспарагиновая кислота, lys – лизин, tyr – тирозин, trp – триптофан.
Для
построения пептидной цепи имеется 20 типов различных «кирпичиков» – природных
аминокислотных остатков. Полипептиды, как известно, составляют основу белков,
выполняющих важнейшие биохимические функции. Эти молекулы имеют набор
механических степеней свободы, которые связаны с флуктуациями около положений
равновесия длин валентных связей, величин валентных углов и с поворотами вокруг
одинарных химических связей. С этими поворотами связаны конформационные
степени свободы, которые ответственны за
многие функции белков и пептидов [3]. Мы не будем в этой статье рассматривать собственно акт химической
реакции. В данный момент нам интересны движения в макромолекуле, которые
подготавливают систему к реакции, обеспечивают изменение структуры после акта
химической реакции (конформационная релаксация), обеспечивают влияние предыдущей
реакции на последующие [3] процессы. Собственно акт химической реакции
описывается в терминах электронно-колебательных и электронно-конформационных
взаимодействий [3], что требует использования аппарата квантовой механики.
Движение системы между химическими актами вполне хорошо может быть описано
уравнениями классической механики при температурах выше 10К. Исключение
составляют атомы водорода и протон, для которых существенны квантовые поправки
даже при 1000К. Эти специальные проблемы мы сейчас также не обсуждаем и
рассматриваем чисто классическую задачу.
В классической механике состояние
системы (наиболее полная возможная информация о системе) из N частиц (атомов) в
момент времени t однозначно
определяется заданием набора векторов координат и скоростей частиц. Обозначим
кратко эти наборы x={xi(t)}и {vi(t)}, которые состоят из 3N чисел (каждый вектор
имеет 3 компоненты). Основная задача механики состоит в том, чтобы найти состояние
системы в произвольный момент времени t, если
известно состояние системы в начальный момент времени (при t=0). В классической механике эта задача решается путем
вычисления траектории движения x(t). Траектория определяется при решении уравнений
движения. В классической механике это
хорошо известные уравнения Ньютона. В нашем случае мы должны написать систему 3N уравнений:
![]() (1)
(1)
![]() - масса атома,
- масса атома, ![]() - соответствующая
проекция суммарной силы, действующей на
атом со стороны остальных частиц:
- соответствующая
проекция суммарной силы, действующей на
атом со стороны остальных частиц:
![]() (2)
(2)
![]() -потенциальная энергия, зависящая от взаимного расположения
всех атомов.
-потенциальная энергия, зависящая от взаимного расположения
всех атомов.
В методе молекулярной динамике
принципиальный вопрос состоит в способе задания U(x). Мы к этому вернемся позднее. Второй принципиальный
вопрос состоит в способе решения системы уравнений движения. Даже для
небольшого белка число уравнений составляет порядка 10000. Основа алгоритма
состоит в следующем. Задав координаты и скорости всех частиц в начальный момент
времени, вычисляют на каждом последующем шаге все силы и новые координаты и
скорости частиц. Расчеты проводят либо при постоянной энергии системы или, чаще,
при постоянной температуре. Мгновенная температура определяется как средняя
кинетическая энергия, приходящаяся на одну степень свободы системы:
![]() (3)
(3)
![]() - постоянная
Больцмана. Температура получается усреднением ее мгновенных значений T(t) по некоторому интервалу времени.
- постоянная
Больцмана. Температура получается усреднением ее мгновенных значений T(t) по некоторому интервалу времени.
Потенциальная энергия молекулы целиком имеет
электромагнитную природу и обычно задается в виде суммы вкладов:
![]()
![]() +
+![]() +
+![]() +
+![]() +
+![]() +
+![]() +
+![]() (4)
(4)
где слагаемые отвечают следующим типам взаимодействий:
![]() - химическим связям;
- химическим связям; ![]() - валентным углам;
- валентным углам; ![]() - торсионным углам;
- торсионным углам; ![]() - плоским группам;
- плоским группам; ![]() - ван-дер-ваальсовым
контактам;
- ван-дер-ваальсовым
контактам; ![]() - электростатике;
- электростатике; ![]() - водородным связям.
- водородным связям.
Эти вклады имеют различный функциональный
вид. Валентные длины поддерживаются за счет потенциала
![]() (5)
(5)
где суммирование проводится по всем химическим связям, ![]() - обозначение для
равновесных валентных длин, r - текущие длины связей,
- обозначение для
равновесных валентных длин, r - текущие длины связей,
![]() - соответствующие
силовые константы.
- соответствующие
силовые константы.
Валентные углы
задаются потенциалами
![]() (6)
(6)
где ![]() - равновесные значения
углов,
- равновесные значения
углов, ![]() - их текущие значения,
- их текущие значения,
![]() - силовые константы.
Энергия торсионных взаимодействий и потенциалов, отвечающих плоским группам,
записываются в одинаковом виде:
- силовые константы.
Энергия торсионных взаимодействий и потенциалов, отвечающих плоским группам,
записываются в одинаковом виде:
![]() (7)
(7)
где n - кратность торсионного
барьера, ![]() - сдвиг фазы,
константы
- сдвиг фазы,
константы ![]() определяют высоты
потенциальных барьеров двугранных углов
определяют высоты
потенциальных барьеров двугранных углов ![]() .
.
Ван-дер-ваальсовые взаимодействия атомов,
разделенных тремя и более валентными связями, описываются потенциалами Леннард-Джонса:
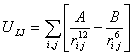 (8)
(8)
Параметры
потенциала A и B зависят от типов атомов i и j, участвующих во взаимодействии; ![]() - расстояние между
этими атомами.
- расстояние между
этими атомами.
Электростатические
взаимодействия задаются кулоновским потенциалом
![]() (9)
(9)
где ![]() ,
, ![]() - парциальные заряды
на атомах,
- парциальные заряды
на атомах, ![]() - диэлектрическая
проницаемость среды.
- диэлектрическая
проницаемость среды.
Водородные связи
возникают и исчезают в процессе движения атомов между теми из них, которые
имеют соответствующий статус. Функциональный вид потенциала водородной связи
похож на потенциал ван-дер-ваальсовых взаимодействий, но с более
короткодействующими силами притяжения:
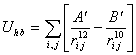 (10)
(10)
Отметим, что система потенциалов (4)-(10) - весьма приближенный способ задания потенциальной энергии. Его недостатки состоят в том, что энергия взаимодействия представляется в виде суммы парных сферически симметричных взаимодействий. И то и другое, вообще говоря, неверно, но с этим приходится пока мириться.
Существуют различные наборы параметров для
потенциалов взаимодействий. Их значения определяются из учета различных типов
экспериментальных данных (спектральные, калориметрические,
кристаллографические) и квантово-химических расчетов. Одна из наиболее
употребляемых систем атом-атомных
потенциалов носит название AMBER и доступна на сайте
www.amber.ucsf.edu
Структура энергетической поверхности полипептида и некондоновские эффекты.
В этом параграфе мы обсудим проблемы,
которым долгое время не уделялось должного внимания. Эти вопросы весьма
существенны и в плане организации деталей расчетной процедуры и для понимания
общих физических свойств пептидных и ряда других систем.
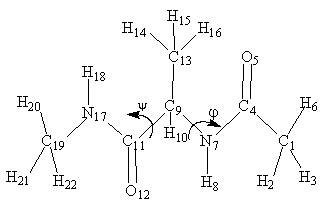
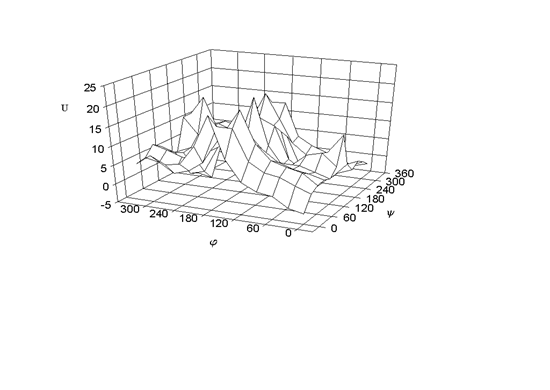 Рис.2. 1- структура модельного
дипептида с двумя существенными конформационными степенями
свободы. Указаны номера атомов, которые используются ниже. 2
– структура энергетической поверхности модельного дипептида. Значение
потенциальной энергии U
(ккал/моль) при каждом значении торсионных углов j и y вычислено при условии
минимизации энергии по всем остальным степеням свободы.
Рис.2. 1- структура модельного
дипептида с двумя существенными конформационными степенями
свободы. Указаны номера атомов, которые используются ниже. 2
– структура энергетической поверхности модельного дипептида. Значение
потенциальной энергии U
(ккал/моль) при каждом значении торсионных углов j и y вычислено при условии
минимизации энергии по всем остальным степеням свободы.
На рис. 2 показана энергетическая
поверхность U(x)
для модельного дипептида. Выбраны только конформационные степени свободы, для которых наблюдаются
наиболее интересные эффекты. Поверхность рассчитана методами современной
квантовой химии [4]. Динамику молекулы
можно уподобить движению частицы по сложному горному рельефу. Имеются локальные
минимумы («ямы»), максимумы («горные вершины») и седловые
точки («перевалы»). Понятно, что путь частицы из одного минимума (конформации) в другой чаще всего лежит через седловые точки (перевалы). Для тетрапептида
количество седловых точек на многомерной
энергетической поверхности составляет порядка 10000. Структура энергетической
поверхности непосредственно влияет на динамическое поведение конформационно лабильных систем и на их функциональную
активность.
![]()
К сожалению, наш мозг так устроен, что мы не
в состоянии наглядно вообразить объекты с размерностью выше 3. Изучение таких
объектов и установление их свойств осуществляется методами дифференциальной
геометрии и топологии (особенно полезна здесь теория Морса) [5]. Если полная
энергия E механической системы
постоянна, то для движения доступны лишь те области переменных {xi(t)}, для
которых E³ U(x) (это следует из не отрицательности значений
кинетической энергии системы). Продемонстрируем особенности геометрической
структуры областей, доступных для
движения.
В жестких молекулярных системах, например,
кристаллах – потенциальная энергия системы может быть представлена в виде суммы
потенциалов типа (5), описывающих сжатие и растяжение валентных связей. Такой
сумме положительно определенных квадратичных членов соответствует многомерный
параболоид: U(x)
~ S xi2 .
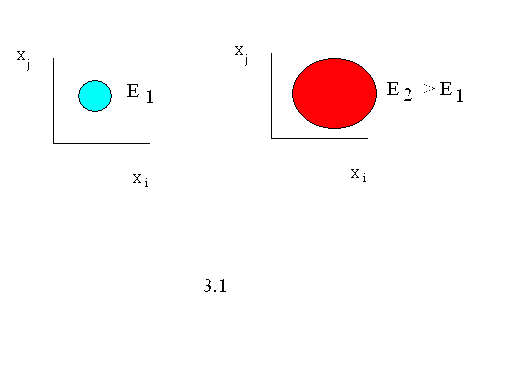
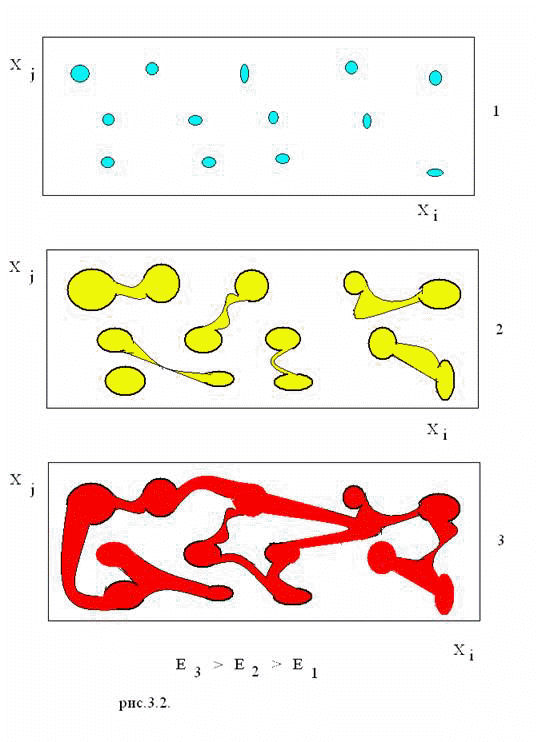 Рис.3. 1 – строение поверхности уровня энергии для
жестких молекулярных систем: поверхность топологически
эквивалентна гиперсфере. Увеличение энергии приводит
лишь к увеличению объема гиперсферы. 2 - строение
поверхности уровня энергии для конформационно
лабильных молекулярных систем. При низких значениях энергии поверхность
распадается на множество несвязанных областей (локусов) (1). Увеличение энергии
приводит не только к увеличению объема этих локусов, но и к возникновению
связей (путей перехода через седловые точки) между
ними (2). Дальнейшее увеличение энергии (3) вызывает увеличение числа и
разветвленности связей между локусами. Увеличение энергии приводит не только к
увеличению объема локусов, но и к топологической перестройке поверхности уровня
потенциальной энергии.
Рис.3. 1 – строение поверхности уровня энергии для
жестких молекулярных систем: поверхность топологически
эквивалентна гиперсфере. Увеличение энергии приводит
лишь к увеличению объема гиперсферы. 2 - строение
поверхности уровня энергии для конформационно
лабильных молекулярных систем. При низких значениях энергии поверхность
распадается на множество несвязанных областей (локусов) (1). Увеличение энергии
приводит не только к увеличению объема этих локусов, но и к возникновению
связей (путей перехода через седловые точки) между
ними (2). Дальнейшее увеличение энергии (3) вызывает увеличение числа и
разветвленности связей между локусами. Увеличение энергии приводит не только к
увеличению объема локусов, но и к топологической перестройке поверхности уровня
потенциальной энергии.
На рис.3.1 представлены области, доступные
для движения в жестких системах при различных значениях энергии Е. Эти области
эквивалентны сферам (гиперсферам) и при увеличении
энергии их объем просто увеличивается.
Совсем иная картина возникает для конформационно лабильных систем, для которых
характерно наличие на энергетической
поверхности не только множества локальных минимумов, но и множества локальных
максимумов и седловых точек (рис.2). При низкой энергии Е область, доступная для
движения разбивается на множество не связанных между собой сфер,
соответствующим локальным минимумам на энергетической поверхности (рис.3.2.1).
С увеличением энергии Е эти сферы увеличиваются в объеме, но до некоторого
предела. Далее, при увеличении энергии,
начинает образовываться сеть из путей, связывающих локальные минимумы по
траекториям, которые проходят через седловые точки. С
увеличением Е эта сеть становится все более и более сложной (рис.3.2.2, 3.2.3).
В природных пептидах значения энергии в седловых
точках, по-видимому, некоторым образом
упорядочено. Это делает их динамическое поведение своеобразным и часто не
похожим на динамику даже ближайших изомеров и гомологов.
К сожалению, мы пока не имеет хорошо
разработанной теории, позволяющей достаточно подробно анализировать свойства
энергетической поверхности пептидов и
белков. Дело здесь не только в математических сложностях. Следующий пример
показывает, что в случае пептидов необходимо существенно совершенствовать и
традиционные представления об электронном строении молекул. Напомним, что
практически вся современная теория молекул основана на приближении
Борна-Оппенгеймера. Основная идея этого приближения проста. Так как масса
электрона в несколько тысяч
раз меньше характерных масс атомных ядер, то скорость движения электрона
примерно на два порядка превышает скорости ядер. Поэтому в первом приближении
можно рассматривать движение электронов в поле покоящихся ядер и не учитывать
влияние динамики атомных смещений на состояние электронов в молекуле. Это
приближение обычно хорошо работает для основного (т.е. самого низкого по
энергии) электронного состояния. Но даже в рамках этого приближения остается
параметрическая зависимость плотности распределения электронов вокруг молекулы
от положений атомных ядер. Обычно этой зависимостью пренебрегают, что
называется приближением Кондона. Для многих жестких молекул это оправдано. Для
пептидов картина совсем другая.
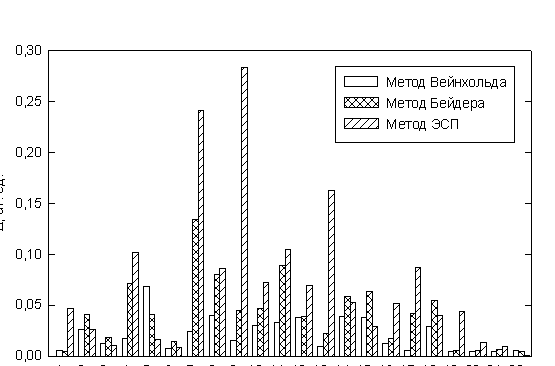 Рис.4. Нарушение приближения Кондона
для пептидов. Максимальные амплитуды изменения атомных зарядов (в единицах
заряда электрона) в модельном пептиде при конформационных
флуктуациях. Номера атомов приведены в соответствии с рис. 2.1. Расчет атомных зарядов проводился тремя
разными методами [4]. Наибольшие эффекты до 0,3e получаются в
рамках метода электростатического потенциала (ЭСП). В соответствии с этим методом заряды на
атомах расставляются таким образом, чтобы распределение ЭСП в окрестности молекулы было как можно ближе к
распределению ЭСП, которое определяется электронной волновой функцией молекулы.
Рис.4. Нарушение приближения Кондона
для пептидов. Максимальные амплитуды изменения атомных зарядов (в единицах
заряда электрона) в модельном пептиде при конформационных
флуктуациях. Номера атомов приведены в соответствии с рис. 2.1. Расчет атомных зарядов проводился тремя
разными методами [4]. Наибольшие эффекты до 0,3e получаются в
рамках метода электростатического потенциала (ЭСП). В соответствии с этим методом заряды на
атомах расставляются таким образом, чтобы распределение ЭСП в окрестности молекулы было как можно ближе к
распределению ЭСП, которое определяется электронной волновой функцией молекулы.
На
рис. 4 показаны изменения интегральной электронной плотности в окрестности
атомов модельного пептида или эффективного атомного заряда, рассчитанного
различными методами при вариации конформации молекулы
[4]. Флуктуации атомных зарядов при конформационных
движениях достигают нескольких десятых единиц заряда электрона. Этот эффект
является одним из проявлений электронно-конформационного
взаимодействия и может вносить существенный вклад в изменение энергии взаимодействия
между группами и в изменение химической реакционной способности. К сожалению, в
настоящее время пока нет возможности в деталях учитывать такого рода эффекты
при компьютерном моделировании динамики молекул
и возможно лишь изучение поправок, возникающих при вариации атомных
зарядов.
Расчет траекторий. Влияние термостата и эргодичность
траекторий.
Задав начальную структуру молекулы и набор
межатомных потенциалов, мы можем приступать к численной процедуре решения
системы уравнений движения и вычисления траектории [6]. Система примерно из
1000 атомов вполне доступна для такого типа расчетов даже на современном
персональном компьютере. Первое, с чем
нужно определиться – с шагом интегрирования, т.е. с минимальным отрезком
времени, на котором мы будем считать приращение координаты частицы
пропорциональным мгновенному значению скорости. Шаг интегрирования должен быть
много меньше характерного времени самых быстрых движений в молекуле – валентных
колебаний. Обычно шаг составляет 1 фемптосекунду (10-15
с).
Расчет обычно проводят при постоянной температуре. В качестве последней понимают среднюю кинетическую энергию атомов (см. (3)). Применяют различные приемы для поддержания постоянной температуры или различные способы задания термостата. Наиболее популярны два типа термостата – термостат Берендсена и столкновительный термостат. В термостате Берендсена вводится для всех степеней свободы сила трения, пропорциональная скорости Fтр.,i = - gvi, которая добавляется к правой части уравнений движения (1). Введение сил трения (или диссипативных сил) - обычный прием в механике для учета рассеяния (диссипации) энергии в тепло при взаимодействии системы со средой. При этом коэффициент трения g строго положителен и пропорционален вязкости среды. В термостате Берендсена коэффициент трения знакопеременный и выбирается в виде g= (mi/t) ((T(t)/T) – 1), где t - характерное время затухания флуктуаций кинетической энергии (обычно порядка 1 пикосекунды), T(t) – мгновенное значение температуры (средней кинетической энергии (3)), T – задаваемая температура термостата. Если мгновенное значение температуры оказывается больше, чем заданное значение Т, то коэффициент трения положителен и энергия отбирается от молекулы. Если ситуация обратная, то g отрицательна и происходит подкачка энергии в молекулярные степени свободы. До недавнего времени термостат Берендсена очень широко использовался при изучении молекулярной динамики белков и пептидов. Однако по мере увеличения производительности компьютеров и переходе к достаточно длинным траекториям (порядка наносекунд) выявилась неприятная особенность этого термостата. Под влиянием термостата Берендсена на достаточно длинных траекториях происходит перекачка энергии от внутренних степеней свободы на поступательное и вращательное движение молекулы как целого. По образному выражению, молекула превращается в стремительно летящий кубик льда (рис.5.1). Не вдаваясь сейчас в физические детали, отметим, что если даже закрепить молекулу, то все равно термостат Берендсена не обеспечивает равнораспределение энергии по внутренним степеням свободы, как это требуют законы статистической физики.
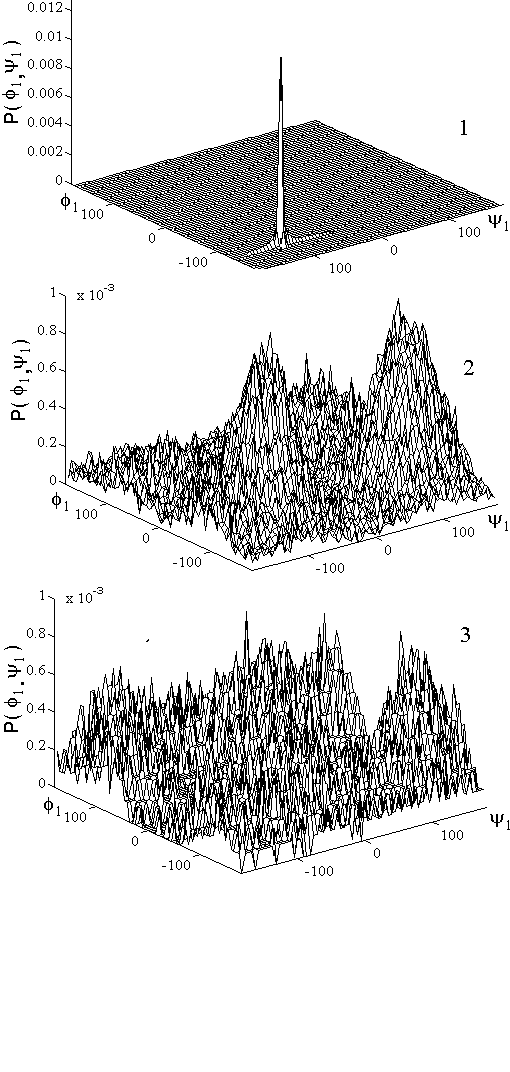 Рис. 5. Двумерные функции распределения
конформационных подсостояний по углам
j и y первого остатка в модифицированном дипептиде asp-asp
(см. рис.1). Области изменения углов от -180о до 180о
даны в линейном масштабе. Длина траектории 5000 пс,
Т=1000К. 1- использован термостат Берендсена (t = 1 пс), центр масс не
фиксирован. Молекула «заморожена» в окрестности одного их минимумов конформационной энергии. Вся энергия перешла в
поступательное движение молекулы как целого. 2 - то же, молекула фиксирована и не может вращаться. Распределение
вероятностей не соответствует результату, ожидаемому из статистической физики. 3 – использован столкновительный термостат. Длина траектории и
температура те же. Среднее число столкновений с каждым атомом 60/пс. Распределение вероятностей близко к результату,
ожидаемому из статистической физики.
Рис. 5. Двумерные функции распределения
конформационных подсостояний по углам
j и y первого остатка в модифицированном дипептиде asp-asp
(см. рис.1). Области изменения углов от -180о до 180о
даны в линейном масштабе. Длина траектории 5000 пс,
Т=1000К. 1- использован термостат Берендсена (t = 1 пс), центр масс не
фиксирован. Молекула «заморожена» в окрестности одного их минимумов конформационной энергии. Вся энергия перешла в
поступательное движение молекулы как целого. 2 - то же, молекула фиксирована и не может вращаться. Распределение
вероятностей не соответствует результату, ожидаемому из статистической физики. 3 – использован столкновительный термостат. Длина траектории и
температура те же. Среднее число столкновений с каждым атомом 60/пс. Распределение вероятностей близко к результату,
ожидаемому из статистической физики.
Более удобным оказалось использование столкновительного термостата. В этом случае каждый атом
молекулы бомбардируют виртуальные частицы,
с максвелловским распределением по скоростям,
соответствующим заданной температуре Т. Задается масса виртуальных частиц
(обычно порядка массы молекулы воды) и средняя частота ударов с каждым атомом
(обычно порядка 50 – 100 ударов в пикосекунду). Взаимодействие виртуальной
частицы с атомом происходит по законам упругих столкновений. Состоянием
виртуальной частицы после столкновения не интересуются. Столкновительный
термостат более эффективно обеспечивает равнораспределение энергии по
внутримолекулярным степеням свободы особенно на длинных траекториях (рис.5.2).
Фактически, столкновительный термостат представляет
собой виртуальный растворитель, динамика которого не важна для исследователя.
Более реалистичный и значительно более
трудоемкий способ поддержания постоянной температуры состоит в учете
взаимодействия молекулы с реальным растворителем, например, водой. В таких
случаях используют системы, содержащие от нескольких сотен до тысяч молекул
растворителя. Преимущества этого метода состоят в явном учете эффектов
растворителя, которые, как известно, весьма существенны для пептидов. Однако
расчеты такого типа относительно редки в виду большой их трудоемкости.
В результате расчета молекулярной динамики
получается траекторный файл, содержащий координаты всех атомов в определенные
моменты времени (обычно запись координат в траекторный файл осуществляют с
шагом 0,1 пс). Это достаточно большой объем
информации. Так, например, для небольшой системы из 50 атомов и при длине
траектории 5 наносекунд такой файл в текстовом формате занимает объем порядка
100 мегабайт. Траекторный файл может быть использован для просмотра
молекулярного кино, т.е. пространственных движений молекулы в процессе расчета.
Такая визуальная информация хотя и полезна, но не может быть использована для
количественного анализа. Количественное изучение динамических свойств молекул
проводят с использованием функций распределения и средних значений от различных
величин. Весьма важно сравнение вычисляемых таким образом значений с
измеряемыми экспериментально величинами. Здесь есть одна тонкость. В
подавляющем большинстве экспериментов измеряемые величины (температура,
давление, характерные времена, амплитуды и т.д.) являются средними значениями
по ансамблю, т.е. по бесконечному множеству точных копий изучаемой системы.
Имея дело с траекторией, мы вычисляем средние значения вдоль траектории, т.е.
по времени. Соответствуют ли средние значения по траектории средним значениям
по ансамблю? Ответ на этот общий вопрос мучительно искал в начале века М.
Больцман. В настоящее время известно, что в случае бесконечно длинных траекторий для
«нормальных» систем ответ на этот вопрос утвердительный. Такие системы
называются эргодическими. В молекулярной динамике мы имеем дело с траекториями
конечной длины. И прежде, чем делать выводы из расчета необходимо убедится, что
траектория обладает достаточно хорошими эргодическими свойствами. В простых
терминах это означает, что система успевает достаточное число раз побывать во
всех доступных областях конфигурационного пространства. Посмотрев на рис.3.2.3
мы поймем, что сделать это за ограниченное время не так просто. Ускорить
процесс преодоления потенциальных барьеров, узких перешейков между локальными
минимумами на поверхности уровня потенциальной энергии можно резко подняв температуру
системы. Например, для дипептидов получение сравнительно хороших эргодических
траекторий требует условий Т ~ 2000К, длина траектории порядка 5 нс (т.е. 5 млн. шагов интегрирования уравнений движения).
Уменьшение температуры приводит к экспоненциальному увеличению длины
траектории, необходимой для достаточно полного сканирования конфигурационного
пространства. При 1750К длина траектории должна быть уже в два раза больше и
т.д. Возникает вопрос, что останется от пептида при температуре 2000К? Ответ здесь
простой. Эта температура является лишь математическим приемом, позволяющим
ускорить прохождение системы через множество потенциальных барьеров и седловых точек. Ни о какой деструкции пептида речи идти не
может, так как валентным связям, задаваемым потенциалами вида (5) разрываться
запрещено при любых температурах.
Корреляционные функции.
Приведем несколько характерных примеров
использования молекулярной динамики для изучения свойств пептидов. Хорошо
известно, что в жестких молекулярных системах (кристаллах) смещение атома около
положения равновесия возбуждает упругие волны скоррелированных
между собой смещений других атомов. Эти коллективные движения (или степени
свободы) называются нормальными колебаниями (модами) или фононами. Выделение
нормальных мод возможно лишь в системах с упругими связями, потенциальная
энергия которых пропорциональна квадрату изменения расстояний между атомами.
Межатомные силы при этом линейно зависят от деформации структуры. В конформационно лабильных системах (например, пептидах)
основная подвижность обеспечивается за счет сильно нелинейных степеней свободы,
связанных с вращением вокруг одинарных связей и с преодолением множества
потенциальных барьеров. Ведение нормальных мод для таких степеней свободы
невозможно. Принципиален, однако, вопрос образуют ли конформационные
степени свободы нелинейные коллективные моды? То есть, существует ли корреляция
между поворотами вокруг отдельных одинарных связей? Этот вопрос интересен не
только с точки зрения физики формирования нелинейных коллективных степеней
свободы. Если коллективные конформационные моды
существуют, то это может иметь непосредственное отношение к функционированию
белков и пептидов. Покажем, как эта проблема решается методами молекулярной
динамики.
В
траекторном файле записана максимально возможная информация о динамике
молекулы. Эта информация сильно избыточная. Поэтому, интересуясь, например,
существованием коллективных конформационных мод нужно
коротко и точно сформулировать вопрос к имеющейся динамической траектории. Конформация пептидной цепи определяется значениями
двугранных углов (рис.1). Однако эти углы определены с точностью до величин
кратных 2p и не
удобны для изучения динамики поворотов вокруг химических связей. При повороте
на угол a меняется
пространственное положение групп. Этому изменению положения групп можно
сопоставить изменение положения точки z(a) на единичной окружности
при повороте единичного вектора на тот же угол (рис.6).
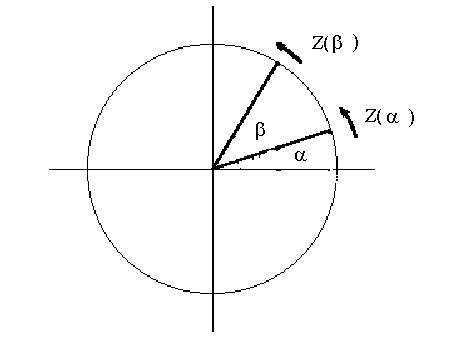 Рис.6. К определению корреляционных
функций для конформационных флуктуаций с участием
двугранных углов. Движение точки Z по единичной окружности (на комплексной
плоскости) сопоставляется изменению пространственного положения групп при
поворотах вокруг химических связей.
Рис.6. К определению корреляционных
функций для конформационных флуктуаций с участием
двугранных углов. Движение точки Z по единичной окружности (на комплексной
плоскости) сопоставляется изменению пространственного положения групп при
поворотах вокруг химических связей.
Если повороты вокруг двух связей по углам a и b происходят независимо, то
среднее значение от произведения <z(a(t+t))z(b(t)> равняется
произведению средних <z(a(t+t))> <z(b(t)>. Знак <…> означает взятие среднего
значения. Напомним, что усреднение проводится по времени t (вдоль траектории). Разность
СС(t) = <z(a(t+t))z(b(t)> - <z(a(t+t))> <z(b(t)> (11)
называется
ниже кросскорреляционной функцией. Эта функция является мерой корреляции (или
взаимозависимости) поворотов по двугранным углам a и b в моменты времени,
разделенными интервалом t. Если (11)
существенно отлично от нуля, то повороты по углам a и b формируют коллективную
степень свободы.
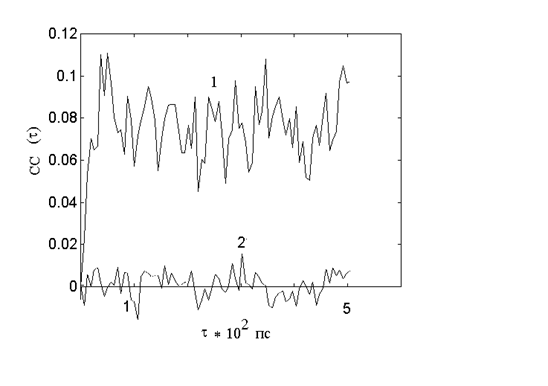 Рис.7. Кросскорреляционные функции
(реальная часть) для пар углов j2c21 (1)
и y2c21
(2) в дипептиде gly-asp (см. рис.1). Из вида кросскорреляционных функций следует, что первая пара углов хорошо коррелирует при движении даже на временах порядка 500 пс. Между второй парой углов корреляция практически
отсутствует даже на малых временах.
Рис.7. Кросскорреляционные функции
(реальная часть) для пар углов j2c21 (1)
и y2c21
(2) в дипептиде gly-asp (см. рис.1). Из вида кросскорреляционных функций следует, что первая пара углов хорошо коррелирует при движении даже на временах порядка 500 пс. Между второй парой углов корреляция практически
отсутствует даже на малых временах.
На рис.7
приведены типичные кросскорреляционные функции для конформационных
степеней свободы. Так, например, в большинстве аминокислотных остатков пара
углов j и c1 оказываются хорошо коррелированной. Однако
это не верно, например, для пары
углов y и c1 (рис.7).
Карты
уровней свободной энергии.
Степень корреляции конформационных степеней свободы тесно связана со структурой поверхности потенциальной энергии. Ниже мы иллюстрируем это обстоятельство с помощью карт уровней свободной энергии пептидов. Рассмотрим это подробнее. Из траекторного файла можно определить относительную частоту или вероятность реализации конформаций молекулы P(a1,a2,…, an). Если при фиксированных значениях двух углов, например, a1 и a2 просуммировать P по всем остальным значениям углов, то мы получим вероятность реализации состояния с определенными значениями переменных a1 и a2. Согласно известной формуле Больцмана, логарифм этой вероятности, умноженный на (-kBT) равен величине свободной энергии данного состояния F. Напомним, что свободная энергия отличается от внутренней энергии U на величину, пропорциональную энтропии S данного состояния: F=U-TS. Энтропия состояния пропорциональна логарифму объема фазового пространства, соответствующего данному состоянию. Чем ниже потенциальная энергия и выше энтропия состояния системы с фиксированными значениями a1 и a2, тем ниже будет соответствующее значение свободной энергии. Таким образом, поверхность свободной энергии (или карта уровней свободной энергии) дает наглядное представление о разделении при заданной температуре областей конфигурационного пространства на доступные и малодоступные области. Доступные области - с низкой потенциальной энергией и относительно большим фазовым объемом. Малодоступные области - с более высокой потенциальной энергией и (или) низкой энтропией. В определенной степени, можно считать, что карта уровней свободной энергии дает интегральную характеристику проекции энергетической поверхности системы на плоскость переменных a1 и a2.
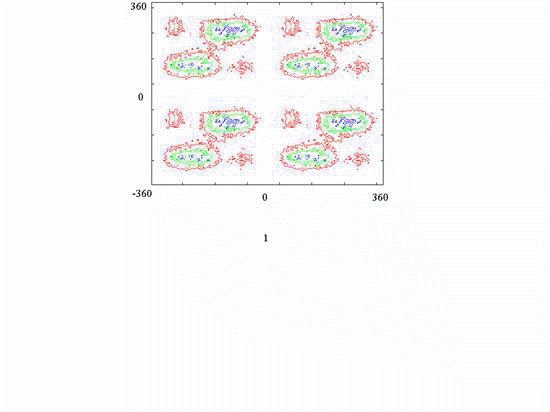
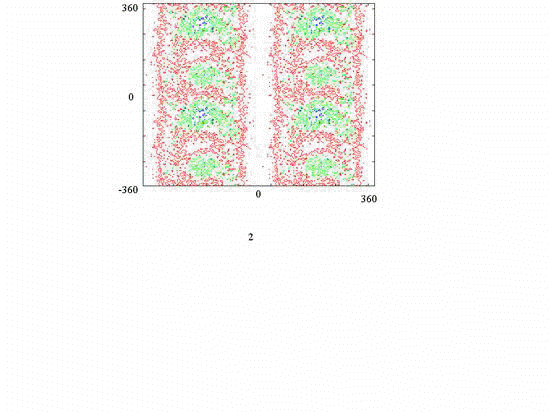
Рис.8. Карты уровней свободной энергии
дипептида gly-asp на плоскостях динамических
переменных j2c21 (1) и y2c21 (2) соответственно.
Для удобства зрительного восприятия диапазон изменения углов расширен до
интервала от -360 до 360 градусов. На карте 1 четко видно два локуса,
соединенных “узким горлом”, что приводит к корреляции динамических переменных
(рис.7.1). На карте 2 наблюдается равномерно шероховатая поверхность
и корреляция отсутствует (рис.7.2).
На рис.8 приведены соответствующие
карты уровней свободной энергии для пар углов, кросскорреляционные функции
которых показаны на рис.7. При этом наглядно выявляется природа формирования конформационных коллективных степеней свободы.
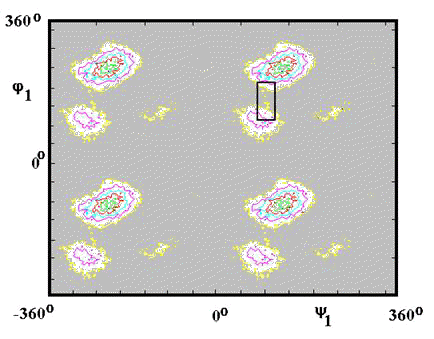
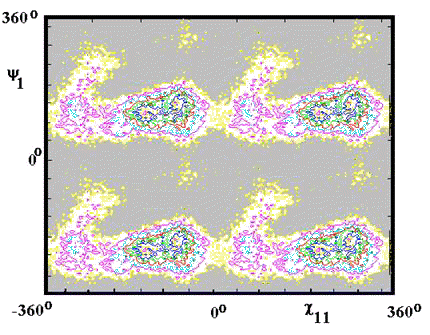 Рис.9. Карты уровней свободной энергии
для модифицированного дипептида tyr-trp (см.рис.1). Области изменения углов от -360 до 360о
в линейном масштабе. Серый фон – область наиболее высоких значений энергии.
Переменная, указанная второй – ось абсцисс. 1 – плоскость (j1,
y1).
Прямоугольником отмечен путь, соединяющий два локуса. Этому пути соответствует
узкое ущелье на энергетической поверхности (см. рис.2). Корреляция ярко выражена.
2 - плоскость (y1,c11).
Широкое изогнутое ущелье - корреляция умеренно выражена.
Рис.9. Карты уровней свободной энергии
для модифицированного дипептида tyr-trp (см.рис.1). Области изменения углов от -360 до 360о
в линейном масштабе. Серый фон – область наиболее высоких значений энергии.
Переменная, указанная второй – ось абсцисс. 1 – плоскость (j1,
y1).
Прямоугольником отмечен путь, соединяющий два локуса. Этому пути соответствует
узкое ущелье на энергетической поверхности (см. рис.2). Корреляция ярко выражена.
2 - плоскость (y1,c11).
Широкое изогнутое ущелье - корреляция умеренно выражена.
Если на карте имеется локусы, соединенные «узким горлом» (рис.8.1., 9.1.)
или имеется сильно изогнутый локус как, например, на рис.9.2), то на взаимные
изменения соответствующих двугранных углов в этих областях наложены ограничения
и возникает корреляция. Если изгибов нет или поверхность равномерно
шероховатая, то данная пара переменных практически независима при движении
(рис.8.2). То есть образование коллективных конформационных
степеней свободы и соответствующее поведение
кросскорреляционных функций непосредственно связано со структурой
энергетической поверхности пептида.
Заключение.
Выше
кратко были изложены основы метода молекулярной динамики и его некоторые
приложения к изучению динамических свойств пептидов. Этот метод очень быстро
развивается вместе с прогрессом в компьютерных технологиях и становится все
более популярным инструментом исследований для молекулярных биологов,
биохимиков, биофизиков и представителей смежных специальностей. В настоящее
время метод молекулярной динамики наряду
с компьютерной молекулярной графикой оказался чрезвычайно полезным при поиске
новых лекарственных препаратов и физиологически активных соединений. Это
направление получило название «молекулярный дизайн». Даже простая
предварительная информация о возможных способах посадки предполагаемой молекулы
- лекарства на соответствующий рецепторный участок позволяет резко сократить
расходы на создание новых фармакологических препаратов. Однако предстоит еще
очень много сделать, прежде чем результаты компьютерного молекулярного
моделирования позволят непосредственно предсказывать функциональную активность
и конструировать молекулярные системы с заданными свойствами.
1. Тихонов А.Н. Молекулярные моторы// Соросовский Образовательный Журнал. 1999. №6. С.8-24.
2. Шипилов А. О дивный новый мир// Компьютерра 1997. №41 (218). С.30-33; Баскин И.
Компьютерное моделирование в молекулярной нанотехнологии// Компьютерра 1997. №41
(218). С. 34-37.
3. Шайтан К.В. Конформационная подвижность белка с точки зрения физики// Соросовский Образовательный Журнал. 1999. №5. С.8-13.
4. Шайтан К.В., Немухин А.В., Фирсов Д.А., Богдан Т.В., Тополь И.А. Электронно-конформационные взаимодействия и значение эффективных зарядов на атомах в пептидах//Молекулярная биология. 1997. Т.31. C.109-117.
5. Фоменко А.Т. Наглядная геометрия и топология. М.: Изд. МГУ, 1998. 223 С.
6. Берлин Ал.Ал., Балабаев Н.К. Имитация свойств твердых тел и жидкостей методами компьютерного моделирования// Соросовский Образовательный Журнал. 1997. №11. С.85-92.
![]()
![]()
![]()