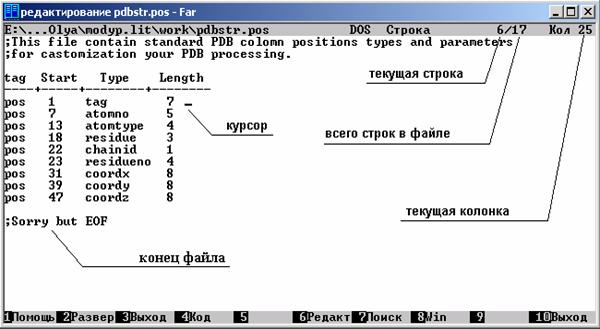
Рис. 9 Файл pdbstr.pos, во второй колонке – номер начальной позиции, в четвёртой – количество столбцов. В третьей колонке: tag – слово "АТОМ", atomno – номер атома, atomtype – имя атома, residue – название остатка, chainid – номер цепи (часто опускается), residueno – номер остатка, coordx, coordy, coordz – координаты по осям x, y и z соответственно.
REMARK periodic box 20 20 20 – размер "ящика" с водой.
АТОМ – указание на то, что далее следует описание атома: уникальный номер атома, имя атома в остатке, название остатка, номер остатка в последовательности и три декартовы координаты атома.
TER – указание на окончание последовательности. Для premd строку нужно удалить.
HETATM – так называемый "гетероатом", то есть атом, не входящий в стандартный для программы, где он был собран, остаток. При запуске программы premd.exe, такие записи игнорируются. Поэтому всегда необходимо заменять слово "HETATM" на "ATOM__" (с двумя пробелами, чтобы сохранить расположение столбцов).
CONECT – описание связей: первый номер – номер атома, который связан с другими (следующие номера). При создании топологического справочника (*.tpl) удобно использовать эту информацию при описании связей.
Файл структуры *.hin
Файл *.hin может использоваться при переводе координат нестандартных молекул в формат str. При этом файлы *.pos и *.tpl не используются. Некоторые параметры, например, правильные заряды на атомах, придётся вносить в *.str вручную.
Пример для молекул, состоящих из стандартных остатков (монопептид пролина в воде):
forcefield amber
sys 0 0 1
view 40 0.065372:
box 20 20 20
seed -1110
mol 1
res 1 ACE 1 - -
atom 1 1H H HC - 0.01 0.5211817 0.8022858 -4.399683 1 2 s
atom 2 CH3 C CT - -0.142 1.401836 0.6444302 -3.778286 4 1 s 3 s 4 s 5 s
atom 3 2H H HC - 0.01 1.760729 -0.3736996 -3.912139 1 2 s
atom 4 3H H HC - 0.01 2.176968 1.339131 -4.09395 1 2 s
atom 5 C C C i 0.616 1.057617 0.8883146 -2.312582 3 2 s 7 s 6 d
atom 6 O O O - -0.504 1.639959 1.773163 -1.677541 1 5 d
endres 1
res 2 PRO 2 - -
atom 7 N N N i -0.229 0.1135035 0.1206233 -1.773465 3 5 s 8 s 18 s
atom 8 CA C CT - 0.035 -0.2958276 0.3186279 -0.4082801 4 7 s 9 s 10 s 12 s
atom 9 HA H HC - 0.048 -0.6812353 1.331479 -0.3078263 1 8 s
atom 10 C C C i 0.526 0.8532816 0.06834622 0.5704045 3 8 s 21 s 11 d
atom 11 O O O - -0.5 1.641413 -0.8649697 0.421405 1 10 d
atom 12 CB C CT - -0.115 -1.411033 -0.6914164 -0.1769076 4 8 s 15 s 13 s 14 s
atom 13 1HB H HC - 0.061 -0.9572152 -1.555701 0.2917716 1 12 s
atom 14 2HB H HC - 0.061 -2.217021 -0.3106433 0.4451269 1 12 s
atom 15 CG C CT - -0.121 -1.908573 -1.079469 -1.565344 4 12 s 18 s 16 s 17 s
atom 16 1HG H HC - 0.063 -2.267472 -2.107143 -1.585348 1 15 s
atom 17 2HG H HC - 0.063 -2.696258 -0.3900914 -1.870854 1 15 s
atom 18 CD C CT - -0.012 -0.684899 -0.8829539 -2.453686 4 7 s 15 s 19 s 20 s
atom 19 1HD H HC - 0.06 -1.003528 -0.5857712 -3.453772 1 18 s
atom 20 2HD H HC - 0.06 -0.1071138 -1.802373 -2.512915 1 18 s
endres 2
res 3 NME 3 - -
atom 21 N N N - -0.463 0.9060994 0.8792239 1.623696 3 22 s 10 s 23 s
atom 22 H H H - 0.252 0.2400249 1.642878 1.637041 1 21 s
atom 23 CA C CT - 0.067 1.340683 0.4332927 2.924588 4 21 s 24 s 25 s 26 s
atom 24 1HA H HC - 0.048 0.4968369 -0.04036276 3.426524 1 23 s
atom 25 2HA H HC - 0.048 2.153537 -0.2852627 2.840401 1 23 s
atom 26 3HA H HC - 0.048 1.649684 1.305584 3.501063 1 23 s
endres 3
endmol 1
mol 2
res 1 WAT 1 - -
atom 1 O O OW - -0.834 -1.791891 1.660319 2.246055 2 2 s 3 s
atom 2 H1 H HW - 0.417 -1.975522 0.7162368 2.264446 1 1 s
atom 3 H2 H HW - 0.417 -2.549447 2.037338 2.708455 1 1 s
endres 1
endmol 2
mol 3
res 1 WAT 2 - -
atom 1 O O OW - -0.834 -1.172214 3.311394 -3.127628 2 2 s 3 s
atom 2 H1 H HW - 0.417 -0.448992 3.608016 -2.55468 1 1 s
atom 3 H2 H HW - 0.417 -1.383347 4.113186 -3.624905 1 1 s
endres 1
endmol 3
Forcefield Amber – тип силового поля – "Amber", только в этом случае можно использовать файл *.hin для перевода в формат *.str.
ATOM – описание атома:
2 – порядковый номер,
CH3 – уникальное имя атома в остатке (для нестандартных молекул в этом столбце прочерк),
C – символ атома в таблице Менделеева,
CT – тип атома в силовом поле,
–0.142 – эффективный заряд,
1.401836 0.6444302 –3.778286 – координаты x, y, z
4 1 S 3 S 4 S 5 S – число и типы связей с другими атомами (s – одинарная, d – двойная и т.д.).
RES – описание остатка:
1 – номер остатка в молекуле,
WAT – название остатка,
2 – порядковый номер остатка.
Описания молекул и остатков создаются по типу вложенных циклов:
mol 1 начало первой молекулы
res 1 ACE 1 - - начало первого остатка
endres 1 конец первого остатка
res 2 PRO 2 - -
endres 2
res 3 NME 3 - -
endres 3
endmol 1 конец первой молекулы
mol 2
res 1 WAT 1 - -
endres 1
endmol 2
Следует обратить внимание, что любая молекула в данном случае содержит остатки. Молекула монопептида состоит из трёх остатков, молекула воды - из одного. Если была собрана нестандартная молекула, то она не будет содержать остатки, их номера и названия надо будет добавить в *.hin вручную! Так выглядит файл для нестандартной молекулы:
forcefield amber94
sys 0 0 1
view 40 0.41209 55 15 1 0 0 0 1 0 0 0 1 -1.7504 0.034747 -55
seed -1111
mol 1
atom 1 - O O - 0 1.75044 -0.8047471 2.221536e-025 1 2 d
atom 2 - C C - 0 1.75044 0.4152529 2.221536e-025 3 1 d 3 s 4 s
atom 3 - H HC - 0 2.685747 0.9552529 -3.306437e-017 1 2 s
atom 4 - H HC - 0 0.8151323 0.9552529 3.306437e-017 1 2 s
endmol 1
Чтобы его можно было переводить в другой формат с помощью программы premd, модифицируем его:
forcefield amber94
sys 0 0 1
view 40 0.41209 55 15 1 0 0 0 1 0 0 0 1 -1.7504 0.034747 -55
seed -1111
mol 1
res 1 UHU 1
atom 1 - O O - 0 1.75044 -0.8047471 2.221536e-025 1 2 d
atom 2 - C C - 0 1.75044 0.4152529 2.221536e-025 3 1 d 3 s 4 s
atom 3 - H HC - 0 2.685747 0.9552529 -3.306437e-017 1 2 s
atom 4 - H HC - 0 0.8151323 0.9552529 3.306437e-017 1 2 s
endres 1
endmol 1
Топологический справочник *.tpl
В файле с расширением tpl содержатся данные, сопоставляющие имена атомов из файла структуры с типами атомов в справочнике силового поля, а также эффективные заряды и расположение связей:
;Residue of Glycine, created by Belyakov A.A.
residue GLY inchain automatic amber96
incoming 1
outgoing 6
tag PDB Type GType Charge Comment
---- ---- ---- ----- ------- --------------
atom N N 3 -0.4157 ;1
atom H H 1 +0.2719 ;2
atom CA CT 4 -0.0252 ;3
atom 1HA H1 1 +0.0698 ;4
atom 2HA H1 1 +0.0698 ;5
atom C C 3 +0.5973 ;6
atom O O 1 -0.5679 ;7
bond 1 2 ;1
bond 1 3 ;2
bond 3 4 ;3
bond 3 5 ;4
bond 3 6 ;5
bond 6 7 ;7
endr ;23.03.01
Каждая запись начинается словом residue и заканчивается словом endr.
RESIDUE – имя остатка (как в файле *.ent), тип остатка, способ вычисления параметров (automatic / manual), тип модели (должен быть тот же, что и после слова mselect в premd.pbatch). Типы остатков: atbegin начальный (первый в цепи), inchain – в цепи, atend – конечный, single – одиночный. Для первых трёх типов остатков указывается номер атома, который соединён с предыдущим остатком (incoming) и / или номер атома, который соединён с последующим остатком (outgoing).
ATOM – имя атома из файла .ent, тип атома из справочника (.ff), количество атомов, с которыми связан данный атом, эффективный заряд. После точки с запятой следует комментарий. В четвёртом столбце нужно указывать именно количество атомов, а не связей. Так, для атома углерода в ацетилене их будет 2. Имена атомов можно перенести из файла *.ent, а типы из *.hin (если мы не забыли преобразовать типы атомов в формат AMBER, когда работали с программой HyperChem!)
BOND 1 2 – между атомами 1 и 2 существует связь. Таким же образом описываются все остальные связи в остатке (без повторов!). Для описания связей удобно пользоваться данными из файла *.ent. Например, строка
CONECT 3 4 56 78 999
будет соответствовать следующим строкам в файле *.tpl:
BOND 3 4
BOND 3 56
BOND 3 78
BOND 3 999
Силовое поле amber *.ff
В этом справочнике содержится информация по всем константам для атомов и групп атомов, которые используются в расчётах. Справочник содержит несколько частей, различающихся формой записи (разделённых пустыми строками):
1. CT 12.01 0.878 sp3 aliphatic C
2. C -CM 410.0 1.444 JCC,7,(1986),230; THY,URA
3. HW-OW-HW 100. 104.52 TIP3P water
4. CT-CT-N -C 1 0.15 180.0 -3. phi,psi,parm94
5. X -X -N -H 1.0 180. 2. JCC,7,(1986),230
6. HW OW 0000. 0000. 4. flag for fast water
7. N NA N2 N* NC NB N3 NT NP NO NY
8. H2 1.2870 0.0157 Veenstra et al JCC,8,(1992),963
1. Атомы:
2. Связи:
3. Валентные углы:
4. Двугранные углы:
5. Псевдоторсионые углы:
6. Водородные связи (потенциал "10-12"):
0000.
. 0000.
7. Эквивалентные атомные типы для параметров Ван-дер-Ваальса. Атомы, следующие после первого, определяются как эквивалентные ему: N NA N2 N* NC NB N3 NT NP NO NY
8. Параметры потенциала "6-12":